Research
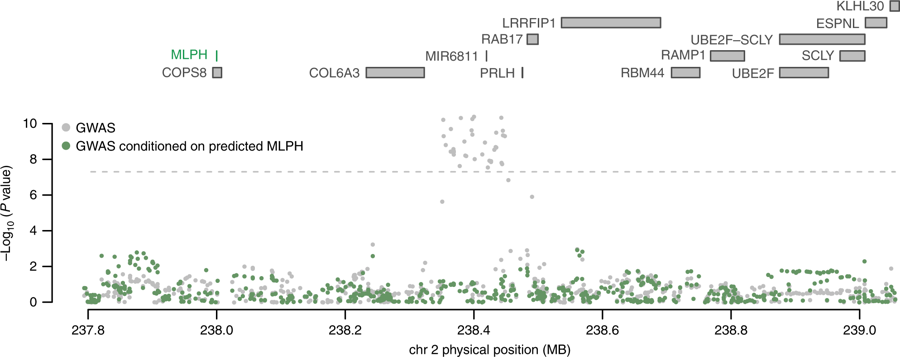
Mechanisms of disease
We are interested in methods that integrate functional genomic data such as gene expression, chromatin interactions, histone acetylation, and chromatin accessibility with GWAS to investigate the genetic basis of complex and monogenic traits. We focus both on novel locus discovery (e.g., by integration of gene expression measurements with association strength to discover new risk genes) and understanding functional mechanisms of disease at known risk loci (e.g., by fine-mapping risk regions from GWAS and overlaying credible causal variants with disease- and cell-type-specific functional features.) Ongoing projects focus on prostate cancer, ovarian cancer, bipolar disorder, and other complex traits. See Mancuso et al., Nat Comms 2018 and Gusev et al., Nat Genet 2018 for examples of our work in this area.
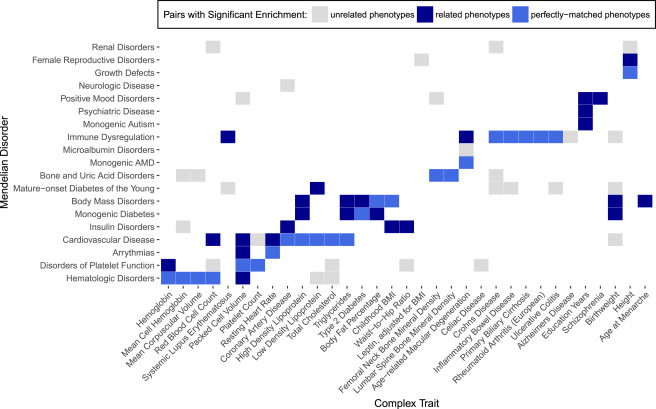
Using genetics and genetic architecture to link traits
Identifying the shared genetic basis between traits, both complex and Mendelian, can illuminate the biological pathways in common. See Freund et al., AJHG 2018 and Jiang et al., Nat Comms 2019 for examples of our work in this area.
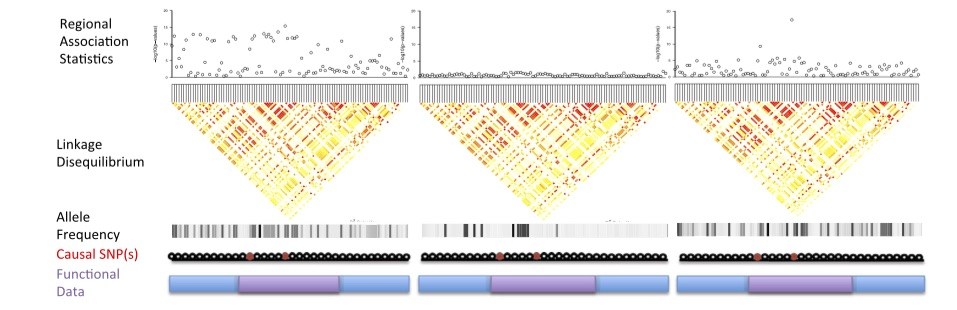
Disease studies in under-represented and admixed populations
Although there exists a wide variety of population-specific genetic variation among humans, the majority of genetic studies and genome-wide assocations studies have been of people of European descent. Our ongoing research focuses on computational modeling of multi-ethnic data to improve disease mapping, involving optimally powered association statistics that take into account differences in the genetic makeup of ancestral populations. For examples of our work in this area, see Gusev et al., Nat Comms 2016 and Kichaev et al., AJHG 2015.